

MOLECULAR GRAPHICS - WEB PROJECTS
The main objective of this project is to gain experience on the use of various tools available for the creation of publication-quality images including but not limited to those of macromolecules. The project will give you a chance to work on your own. What you will learn in class are the basic features of the software and at the end it is each student's creativity that will determine the quality of the report.
Each group will be assigned a different protein-ligand complex identified by a PDB code. Keep in mind that specific parts of the sequence are the target of research because they are functionally the most important to the protein. Thus you can simplify the complexity of your investigation by restricting the search space to only the residues near the target site. Here, 'near' is best if it refers to spatial distance, which is not always related to distance in the amino-acid sequence.
-Regarding file storage / Computer Usage
Remember that the files that we make in class do not have to be the ones you will use in your paper. Your atomic coordinate files (.PDB, .CHM) can be stored on a floppy disk, if you need to transport files larger than ~1.44 MB you can e-mail them to yourself. The main idea is NOT to become dependent on the computer that you will be using in class, since access to them is limited.
Unfortunately, we do not have access to the administrator privileges on the lab computers. To bypass that problem, your instructor will make a folder [Stand_Alone] available for you to copy and paste in the computers desktop. This folder contains stand_alone programs, that is they run without installing them in the hard drive.
The RayTracer and VMD cannot be made into stand_alone executables. If you need to install these programs ask your instructor how to do it, be advised that the program might not work after log off and all saved work will be lost.
PROCEDURE
Retrieve the atomic coordinates of the protein
i. Go to the BROOKHAVEN PROTEIN DATABASE website
ii. In the SEARCH THE ARCHIVE box enter your PDB code.
iii. Click on DOWNLOAD / DISPLAY FILE. In the DOWNLOAD THE STRUCTURE FILE section, click on the X in column #1 and row #1. This will give you access to the uncompressed PDB file. The files go to a DEFAULT directory when you download them, make sure you know where the files are going.
Getting started
As your first task, you will locate the main text reference. To do that open the assigned PDB file in a text editor [we have WORDPAD in the computers]. The main reference is listed in the line starting with JRNL
After reading the main reference paper, prepare an outline:
i. Protein function, ligand identity and any required cofactors
ii. The residue(s) of interest. These are identified by number within the sequence.
iii. You will need to cite other papers as well. Include at least one REVIEW ARTICLE, these type of article contains most of the background information available up to its publication date, the references cited are about all you will need to know to write your introduction and get an idea as to what kind of pictures you will want to include on your report.
View the protein
We will use RASMOL, Swiss-PdbViewer, and MolMol. Don't worry if you only know a couple of commands by the end of the class, you will learn to use the programs by using them. Two common functions to know about as soon as you retrieve your PDB file are:
i. [Hold LEFT CLICK, then move]: rotates the protein so you can view them from different angles.
ii. [Hold LEFT+RIGHT CLICK, then move up/down]: Will zoom the protein IN/OUT. You will be required to take some snapshots of the target site and describe what you see.
At this point we should get to know what the FILES really are, let's take a look at the fourth line of this PDB file:
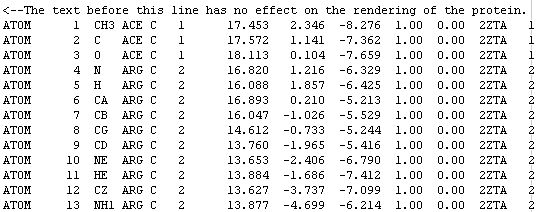
The word ATOM identifies the line of text as an atomic coordinate data set. The molecular renderers will not display an atom unless the line starts with the word ATOM (or HETATM for special atom types).
The number 4 means this is the fourth atom in the sequence.
N stands for main-chain Nitrogen; other atoms are identified with their positions within the side-chain. For example, atom number 6 is an alpha carbon and atom number 7 is a beta carbon.
ARG C means this atom belongs to an arginine residue of subunit C. Some proteins have many subunits.
The number 2 is the residue number. ACE is a neutralizing cap and has assigned the number 1, but most of the time it is assigned the number 0.
The three numbers [16.820 1.216 -6.329] are the [X Y Z] coordinates of the atom. The units are ANGSTROMS.
-Cleaning up the file
As you can see, part of the file is not needed. The crystal structure also includes water molecules, which make viewing difficult in certain display modes. What you actually need are the lines that start with the word ATOM.
If you want to show only a few residues in one of your pictures, you can delete all residues except for the ones you are interested in. Alternatively, you can use the restrict command in the RASMOL viewer. For example restrict 12, 13, 26 will make all except residues 12, 13, and 26 dissapear from view.
-Mutation guidelines
You might want to make a figure showing how a residue change [mutation] will affect the function of the protein.
i. OPTIONAL: go to the SWISS VIEWER tutorial page.
ii. Follow the instructions from your individual assignment.
iii. Open the SWISS VIEWER and close the splash screen. For some reason it does not close on its own.
iv. You can load the file by the drag-n-drop method. For some file types you will need to go to FILE, OPEN PDB FILE.
v. The mutation process is very simple and employs a dropdown menu of the available amino-acids. The next step is more complex and you are required to choose a CONFORMER. If this is not mentioned in the pre-lab, ask your instructor about guidelines for choosing the best conformer. (More material for your Introduction / Abstract)
vi. Use descriptive names for your files. For example, suppose you downloaded the file 4XVF.pdb and you mutate ASN at position 25 to LEU, then you might call your file 4XVF_N25L.pdb
You might want to perform an Energy Minimization if you have replaced side-chains in a PDB structure. Comparison of the energy of a number of models is helpful for selecting the best one. You could submit 2 files with different torsion angles for the residue that was replaced; the correct torsion will result in the lowest energy structure. After you have your PDB files ready, submit them to the EEF1 SERVER a few minutes later, you can check your e-mail for the minimization results.
All you need are the new atomic coordinates and the number of atoms, this protein has 325 atoms. Some molecular renderers will not display the protein unless this number is included in the file. Just follow the link and when the page opens click on Coordinate file after minimization. What you will see is a page full of text:
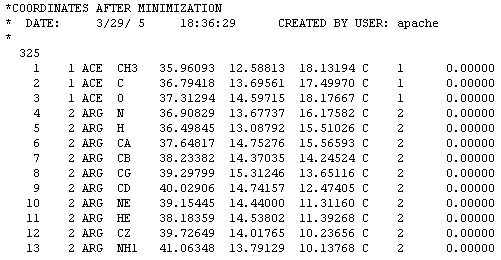
This file does not look like a PDB file because it is written in CHARMM format. Don't worry about that. Just COPY from the web page and PASTE to a text editor such as NOTEPAD. Then save the file and add the CHM extension to identify it as a CHARMM file. The file can be displayed in the RASMOL viewer as is. For use in other applications where a PDB will work better we need to convert it to PDB format. Some protein viewers let you open this file format and SAVE AS PDB.
REPORT FORMAT
Unlike most reports, this one has to contain little text and mostly pictures. The pictures in your report have to include a caption explaining what the figure is and what software you used to create it. For example:
MINIMUM REQUIREMENTS
-A short introduction with background information about the protein, the best source is a review article.
-One sequence alignment of the protein sequence and a homologous one. Use a web-based program to align two of your sequences, show a snapshot of the sequence alignment. You do not have to align the entire protein sequence, only the residues where the active site is located is of interest to us.
-One Picture comparing the ligand when bound to the protein and in the free state. You could use the MAGIC FIT function in the SWISS-VIEWER to align the two structures.
-One picture of the ligand and the residues where it binds. Display only those atoms in STICK mode, the rest of the protein should not block the view. A good way to do this is by using the RESTRICT command in RASMOL or in the SWISS-VIEWER by turning all residues off and then turning on only the ones you want. Or better yet the residues that are not envolved in ligand binding could be displayed as strands, thin ribbons or be made transparent.
-At least one POV-RAY picture of the protein. To get the starter file open the PDB file in a viewer and save as POV-RAY scene. Programs that do that include the SWISS-VIEWER and VMD. If you want to get fuzzy with the pictures, change the camera coordinates, light intensity, texture...
-At least one picture using POV-RAY objects, primitive or otherwise, for example

-At least one composite picture. Meaning you are to glue various images together, for example
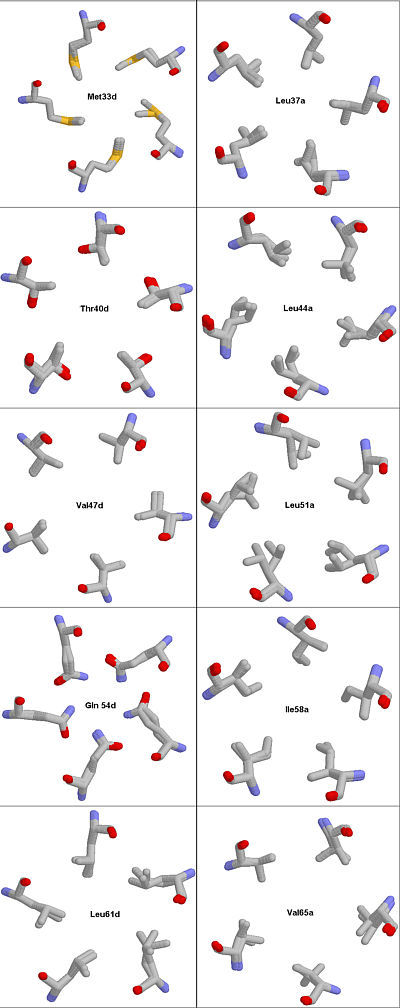
Fig. #. Structure fitting of 1MZ9 and 1VDF. Because the core residues assume different rotameric states when the protein is complexed with vitamin D3 as compared to the uncomplexed COMP the MAGIC FIT utility of the SWISS VIEWER cannot fit all atoms from 1VDF into the 1MZ9 model.
Rendered with RASMOL.
-Include data for at least one task performed by using a web-based server (besides the sequence alignment). This could use Energy Minimization, Homology Modeling, Ligand Docking...
-REFERENCES to research papers where you found information and ideas for your pictures. For that we will do a LITERATURE SEARCH.
USEFUL LINKS
[POV-RAY]_
[Swiss-PdbViewer]_
[JUN]
[PyMOL]_
[RASMOL]_
[MolMol]_
[VMD]
EEF1 SERVER: The server will take your PDB-format coordinates and perform 300 steps of energy minimization using EEF1 and the ABNR method. Comparison of the energy of a number of models is helpful for selecting the best one. You will also want to perform energy minimization if you have replaced side-chains in a PDB structure.
The Dundee PRODRG2 Server: This WWW PRODRG server will generate energy minimized coordinate files of small molecules. You can use this for the ligands in your PDB file.
ExPASy Proteomics tools: Tools from ExPASy and other servers.
The Online POV-Ray Tutorial: Excellent resource for POV-Ray users, from novice to expert. It is well designed and documented.