

Physical Chemistry Laboratory [CHEM 335]
Structure and Biological function of the Cartilage Oligomeric Matrix Protein
[Molecular Graphics - Web Projects]
Serena Blocker♥, Sian Ramos♦ and Jin Rei♦♣
ABSTRACT
This report aims to illuminate that side of the Cartilage Oligomeric Matrix Protein (COMP) that is only available through techniques used in molecular modeling and computational chemistry. We start with some background information about the protein and build a model of the biologically active form. Then we use more advanced molecular modeling software to highlight the parts that are functionally important to the protein and provide as much instructions as seems appropriate to enable the reader to generate his/her own publication-quality images. We also make use of resources available in the World-Wide Web and examine the molecular modeling literature.
♥Corresponding Author
♦Contributed similarly to this work
♣Computer modeling/Raytracing expert
INTRODUCTION
The Cartilage Oligomeric Matrix Protein (COMP) is a multidomain protein belonging to the thrombospondings family and is designated type TSP-5 [1]. Electron Microscopy images of native state COMP issolated from the Swarm rat chondrosarcoma have revealed bouquet-like structures with a central coiled coil assembly and five flexible linkers that connect to the cell surface [2].
The biological role of COMP is not very well stablished but it is believed to play an important role in human cartilage because mutations of the protein lead to premature arrest of bone growth and joint laxity [3]. The binding capacity of COMP for vitamin D3 has been reported [4]. The best-known function of vitamin D3 as the primary precursor needed for cartilage and bone mineralization together with the predominance of COMP in the proliferative region of developing cartilage suggests a storage function for the protein.
METHODS AND RESULTS
In order to obtain good crystals, the proteins have to be free of large flexible domains. The structure with PDB code 1MZ9 is the coiled coil domain of COMP, this is the most rigid part of the protein and is the one most likely to generate crystals. Images of the entire protein can be obtained by electron microscopy. Based on the specifications of Morgelin et al [2], we constructed a model using SWISS VIEWER and POV-RAY (Fig. 1). The thin ribbons were generated from a modified version of the PDB file 1MZ9. This file contains frayed C-termini beyond residue # 30 on all five helices. The PDB was displayed in the SWISS VIEWER in ribbon display mode with all residues turned off. Flexible linkers are not drawn to scale. The five subunits comprising the coiled coil domain were enclosed in a cylindrical surface ~3 nm in diameter using the cylinder command with the open keyword in POV-RAY. The cylinder was given a glass texture and certain value of transparency, that is why the helices of the coil are vissible. The cell surface is an infinite plane. The globular domains that grab the cell surface were drawn with the cone command.

Fig. 1. Model of Cartilage Oligomeric Matrix Protein bound to a cell surface.
Rendered with SWISS VIEWER and raytraced with POV-RAY.
It has been found that vitamin D3 is required for cartilage and bone mineralization. The vitamin is insoluble and its transport and storage is made possible by vitamin D-binding proteins as of yet not well characterized. Coincidentially, the pattern of hydrophobic core residues in COMP provide a favorable mechanism of interaction with vitamin D3 and the crystal structure of such complex is available in the PDB. The vitamin-binding capacity of COMP provides an attractive model for the cellular storage and delivery of insoluble vitamins.
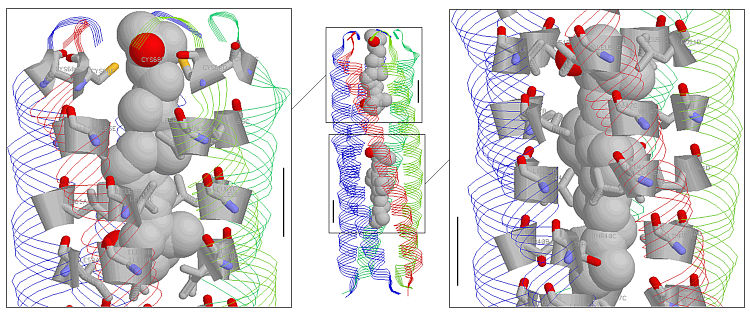
Fig. 2. COMP in complex with two molecules of vitamin D3 (shown as van der Waals spheres). The insets show a close-up view of the core residues that interact with the vitamin molecules, these are shown in sticks mode. The scale bar is 6.0 Angstroms.
Rendered with RASMOL.
In order to get a clear view of the vitamin D3 binding mode we displayed PDB file 1MZ9 in the RASMOL viewer (Fig. 2). The central figure shows the overall structure and was obtained using the strands display mode and color by chain. The vitamin molecules were displayed using the select 1001,1002 command, where the numbers indicate the numbering of the vitamin molecules in the PDB file. The color mode for the vitamins is CPK. For the close-up views we issolated (by clicking and looking at the command line window) the residues that pointed towards the core of COMP. These are residues #30, 33, 37, 40, 44, 47, 51, 54, 58, 61, 65, and 68. The protein folds as a coiled coil and the residues are denoted by heptad repeats abcdefg where the a's and d's point towards the core of the structure.
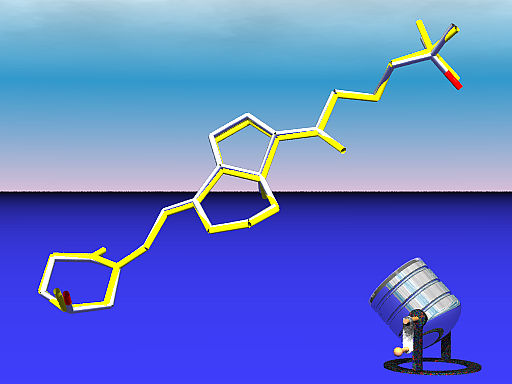
Fig. 3. Vitamin D3 molecule I from PDB file 1MZ9 (shown in CPK) aligned with energy-minimized model (shown in yellow) built by the Dundee PRODRG2 Server [5]. The fluorescent lamp was added because we wanted to get fuzzy with our graphics.
Rendered with SWISS VIEWER and raytraced with POV-RAY.
Ligands tend to change conformation upon binding and the vitamin molecules bound to COMP are likely to assume a conformation that is very different from the free molecule. We reasoned that the structure of free vitamin D3 molecule could be approximated by an energy-minimized model. We made a PDB file with the atomic coordinates of the vitamin D3 bound to the N-terminus of COMP and aligned the structure with the one built by the Dundee PRODRG2 Server [5]. The structures were aligned using the SWISS VIEWER and saved as a POV-RAY scene. Each molecule was given a distinc color and a transparent glass texture. That way, none of the molecules blocks each other's view.
The alignment option was all atoms. The fact that the molecules are not superimpossable indicates that in the bound state, vitamin D3 adopts a slightly unfavorable conformation in order to fit into the COMP channel (Fig. 3).
We used the MAGIC FIT utility of the SWISS VIEWER to investigate global changes in the COMP structure. For that purpose we downloaded the PDB file 1VDF, which corresponds to uncomplexed COMP (exept for a chloride ion at the GLN54d constriction). The two structures were aligned in the SWISS VIEWER and saved as a PROJECT which was then displayed in RASMOL.
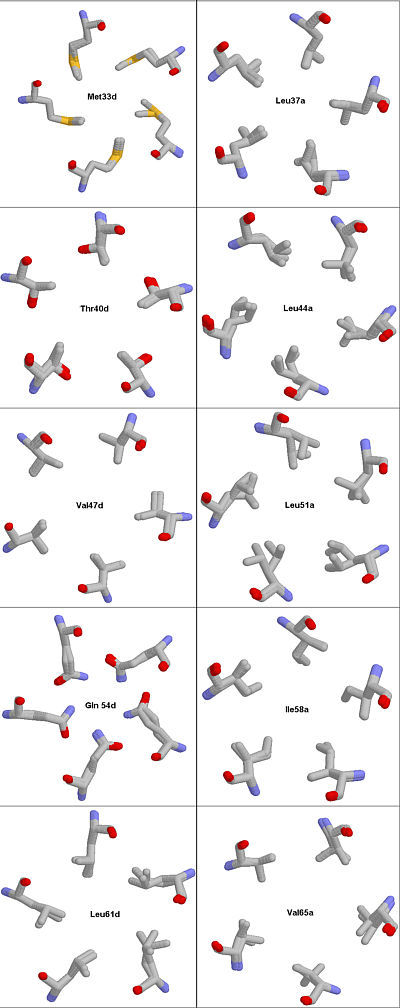
Fig. 4. Structure fitting of 1MZ9 and 1VDF. Because the core residues assume different rotameric states when the protein is complexed with vitamin D3 as compared to the uncomplexed COMP the MAGIC FIT utility of the SWISS VIEWER cannot fit all atoms from 1VDF into the 1MZ9 model.
Rendered with RASMOL.
The backbone coordinates of the 1VDF file fitted quite well into the 1MZ9 model but the side chains did not. It is common for solvent exposed side chains to exhibit large deviations from structure to structure. However, the core positions are restricted to a narrow range of conformational space. Changes in dihedral bonds at these positions are inherent to steric effects from ligand binding. To get a better idea as to the magnitude of the deviations in dihedral angle values we used the SET PICKING TORSION command in the RASMOL viewer. Table 1 shows the results of long hours of clicking and reading the COMMAND LINE.
DIHEDRALS FOR HELIX A OF 1MZ9 ------------------------------------ RESID CHI1 CHI2 CHI3 MET33d 87.61 -66.92 -179.99 LEUa37 -77.79 -67.12 0.00 THR40d -68.17 0.00 0.00 LEU44a -94.91 -174.78 0.00 VAL47d -44.36 0.00 0.00 LEU51a 171.04 -172.45 0.00 GLN54d 66.17 -171.56 -175.81 ILE58a -70.55 -61.86 0.00 LEU61d -55.79 -170.44 0.00 VAL65a -79.06 0.00 0.00 CYS68d -81.46 0.00 0.00 |
DIHEDRALS FOR HELIX A OF 1VDF ------------------------------------ RESID CHI1 CHI2 CHI3 MET33d 76.37 -67.56 178.66 LEUa37 -78.87 -66.33 0.00 THR40d -68.31 0.00 0.00 LEU44a -101.32 -69.52 0.00 VAL47d -45.26 0.00 0.00 LEU51a 54.84 -69.25 0.00 GLN54d -29.00 -169.17 -177.38 ILE58a -41.81 -61.90 0.00 LEU61d -56.18 -170.14 0.00 VAL65a -81.70 0.00 0.00 CYS68d -63.47 0.00 0.00 |
Table. 1. Dihedral angles for helix A of the COMP protein. These show that the core residues assume different rotameric states when the protein is complexed with vitamin D3 as compared to the uncomplexed COMP. Angles where obtained with RASMOL using the SET PICKING TORSION command.
Having the least steric constraints, structure 1VDF should have its core side chains in the minimun energy conformation. We used the EEF1 server to minimize and measure the energy of helix A from 1VDF and 1MZ9 [6]. Energy minimization results are meaningful if the two structures have the same number of atoms, thus we removed a residue (GLY46) from 1VDF because 1MZ9 is truncated at CYS45. The results are shown in Table 2. Note that the values refer to one helix only, thus the difference in stability is about five times as large as the table shows.
| 1MZ9 Total Energy | Bonds | Bond Angles | Dihedral Angles | Improper Torsions | Van der Waals | Electrostatics | Solvation Free Energy |
| -1404.0 | 8.8 | 32.4 | 21.0 | 5.6 | -272.5 | -720.7 | -478.4 |
| 1VDF Total Energy | Bonds | Bond Angles | Dihedral Angles | Improper Torsions | Van der Waals | Electrostatics | Solvation Free Energy |
| -1406.6 | 8.8 | 34.3 | 26.4 | 6.0 | -278.3 | -735.0 | -468.8 |
Table. 2. Energy minimization results obtained with EEF1 server for helix A of the COMP protein. The results show that the protein is slightly more stable when it is not complexed with vitamin D3.
Experimental data suggest that binding of hydrophobic ligands is energetically favourable [4]. However, we find that the protein is lower in energy when it is not complexed with vitamin D3. Our results are not at all contradicting experiment. What we see is that the core residues assume energetically unfavorable conformations to fit the ligands and the complexed COMP is higher in energy than the uncomplexed form in a per helix basis. These effects are probably counterbalanced by favourable van der Waals interactions of the core residues with the ligand.
Since the two vitamin D3 molecules fit into non-equivalent constrictions, we reasoned that their conformations would be different. We made a PDB file using the SELECT 1001, 1002, 54 command in RASMOL then we saved the file using SAVE PDB D354, where D354 is the filename. The file was displayed in VMD and rendered as a POV3 scene. The final result of our work with vitamin D3 is shown in Fig. 5. We decided to get fuzzy again and placed the molecules hovering above calm water. The 1MZ9 structure was missing the chloride ion that complexes with GLN54d in 1VDF so we rendered the electron density of the ion as a wooden sphere.
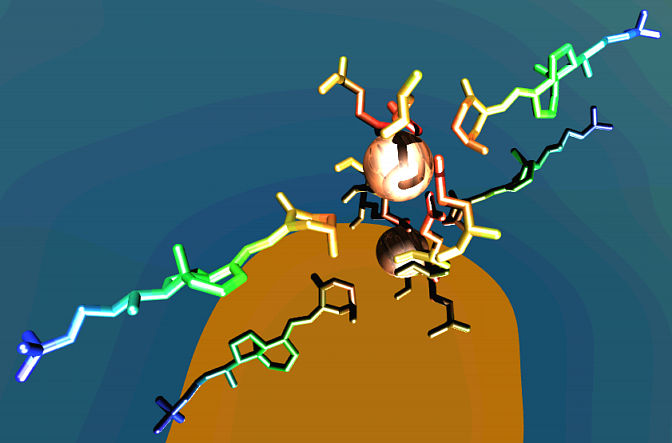
Fig. 5 The two vitamin D3 molecules (molecule I at the left and II at the right). The two cavities in COMP are separated by the conserved residue GLN54d. The electron density of the chloride ion that binds GLN54d in the 1VDF structure has been rendered as a wooden sphere.
Rendered with VMD and raytraced with POV-RAY.
Our vitamin D3 rendering shows that the two molecules look very similar even though the COMP channel is large enough to accomodate different conformations of the steroid ring and the long tail. The conformation exhibited by the bound vitamin D3 is very similar to our energy minimized model so we can conclude that the bound and the free ligand have nearly the same conformation.
DISCUSSION
In this study we have examined the structural features of COMP bound vitamin D3 and in its unbound form. Our work highlights the impact of computer molecular modeling in illustrating the structure and function of macromolecules. The results of our work leads to the following conclusions: [a] while a graphical user interface makes the creation of computer graphics quite easy, it is also very easy to ruin a project in a single click, [b] it is very important to know what parts of the protein to display in order to convey meaning and [c] you can never get too fuzzy with your graphics.

Fig. 6. Sequence alignment of the coiled coil domains of COMP from rat and human.
In a closing note, we compared the amino acid sequence of the coiled coil domain of COMP from rat and human. The alignment was done using T-COFFEE [7]. The results show that the two sequences are nearly identical, differring in only three out of forty six residues. This means that rats ands humans are not so different after all.
BIBLIOGRAPHY
[1] Adams J "Thrombospondins: multifunctional regulators of cell interactions" Annu Rev Cell Dev Biol, 2001: 17, 25�51
[2] Morgelin M, Heinegard D, Engel J, Paulsson M "Electron microscopy of native cartilage oligomeric matrix protein purified from the Swarm rat chondrosarcoma reveals a five-armed structure" J. Biol. Chem. 1992: 267, 6137-6141
[3] Marti C, Neidhart M, Gerber T, Hauser N, Michel BA, Hauselmann HJ "Cartilage Oligomeric Matrix Protein (COMP): Die Rolle eines nicktkollagenen Knorpel-Matrix-Proteins als Marker der Krankheitsaktivitat und Gelenkzerstorung bei Patienten mit rheumatoider Arthritis und Arthrose" Z. Rheumatol, 1999:58:79-87
[4] Ozbek S, Engel J, Stetefeld J "Storage Function of cartilage oligomeric matrix protein: the crystal structure of the coiled coil domain in complex with vitamin D3" The EMBO Journal, 2002: 22, 5960-5968
[5] Schuettelkopf A, van Aalten D "PRODRG - a tool for high-throughput crystallography of protein-ligand complexes" Acta Crystallographica 2004: D60, 1355-1363
DUNDEE Server URL:http://davapc1.bioch.dundee.ac.uk/programs/prodrg/
[6] Lazaridis T, Karplus M "Discrimination of the native from misfolded protein models with an energy function including implicit solvation" J. Mol. Biol. 1999: 288, 477-487
EEF1 server URL:http://mingus.sci.ccny.cuny.edu/server/
[7] Notredame C, Higgins D, Heringa J "T-Coffee: A novel method for multiple sequence alignments" Journal of Molecular Biology 2000: 302, 205-217
URL:http://www.ch.embnet.org/software/TCoffee.html
[8] MERCURY'S HELP DESK | MOLECULAR GRAPHICS - WEB PROJECTS
URL: http://jr.stryker.tripod.com/physchem/graphics.html (Retrieved April 06 2000)
[9] Humphrey, W., Dalke, A. and Schulten, K., "VMD - Visual Molecular Dynamics", J. Molec. Graphics, 1996: 14, 33-38
URL:http://www.ks.uiuc.edu/Research/vmd/
Sayle R, Milner-White E "RasMol: Biomolecular graphics for all", Trends in Biochemical Sciences (TIBS), 1995: 20(9), 374
ULR:http://www.umass.edu/microbio/rasmol/
[10] Guex N, Peitsch, M "SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling" Electrophoresis 1997: 18, 2714-2723
URL:http://www.expasy.org/spdbv/
[11] POV-RAY:Persistence of Vision Raytracer, URL:http://www.povray.org/
APPENDIX: RAW DATA/NOTES
Main Citation:
The EMBO Journal, 2002: 22, 5960-5968
PDB code of protein: 1MZ9
Structure of 1MZ9 is the coiled coil domain of COMP. It exhibits five-fold symmetry.
TEM pictures of whole COMP at:
J. Biol. Chem. 1992: 267, 6137-6141
1MZ9 contains two vitamin D3 molecules # 1001 and 1002
Core residues hold the ligands and are denoted "a" and "d"
MET33d, LEUa37, THR40d, LEU44a, VAL47d, LEU51a, GLN54d, ILE58a, LEU61d, and VAL65a
RASMOL COMMANDS/FUNCTIONS:
HOLD LEFT CLICK AND MOVE --> ROTATE
HOLD RIGH CLICK AND MOVE --> TRANSLATE
HOLD SHIFT + [RIGHT+LEFT CLICK] AND MOVE UP --> ZOOM OUT
HOLD SHIFT + [RIGHT+LEFT CLICK] AND MOVE DOWN --> ZOOM IN
save pdb FILEMANE --> save selected atoms to a PDB formatted file
default directory to save file is RASWIN [WIN = WINdows]
select a directory where you can find the file
set picking...
distance --> click two atoms
angle --> click three atoms
torsion --> click four atoms
restrict...
1,2,3 --> makes all residues except 1, 2, and 3 dissapear from view
HOH --> makes all everything except water dissapear from view.
try other residue or molecule names in the PDB file.
select...
1,2,3 --> commands, display modes, etc. apply only to selected entities.
SEQUENCE of rat and human COMP in one-letter code FASTA format
>RAT
GDL APQMLRE LQETNAA LQDVREL LRQQVKE ITFLKNT VMECDAC G
>HUMAN
SDL GPQMLRE LQETNAA LQDVREW LRQQVKE ITFLKNT VMECDAC G
POV-RAY RELATED URLS:
http://www.povcomp.com/hof/
http://library.advanced.org/3285/tutorial/simple.html
BUILD AND MINIMIZE LIGAND AT THE DUNDEE SERVER:
http://davapc1.bioch.dundee.ac.uk/programs/prodrg/